Introduction
리증후군(Leigh syndrome)은 미토콘드리아성 산화인산화과정(oxidative phosphorylation, OXPHOS)에 장애가 발생하는 신경퇴행성 질환이다[1]. 이는 소아에서 가장 흔한 사립체유전질환으로 정상 출생아 34,000~77,000명 당 1명의 빈도로 발생한다고 알려져 있다[2]. 리증후군은 임상증상, 신체진찰, 생화학적검사, 그리고 뇌자기공명영상을 통해 진단할 수 있는데, 리증후군의 임상양상은 환자마다 큰 차이를 보일 수 있어, 증상의 발현 시기, 중증도 및 예후가 매우 다양하다[3,4]. 대부분의 환자들은 생후 3~12개월의 영아기에 증상이 시작되고, 발달단계의 퇴화, 운동실조, 근긴장감소 등을 보이면서, 뇌간기능장애로 진행한다[3,4]. 하지만, 청소년이나 성인에서 증상이 발현되기도 하고, 신경학적인 증상없이 영아 돌연사증후군으로 사망 이후, 부검을 통해 리증후군으로 진단되는 경우도 있다[3,5].
저자들은 심정지를 첫 증상으로 보인 리증후군 영아에서, 새로운 NDUFS1 돌연변이를 확인하였기에 보고하는 바이다.
Case description
생후 22일된 남아가 심정지로 응급실에 내원하였다. 내원 1일전부터 35.5~35.8도의 저체온을 반복적으로 보였고, 내원 당일 얼굴이 창백해 보이면서 처짐 소견으로 구급차통해 응급실에 도착하였다. 도착당시 자발호흡이 없는 심정지 상태로 심폐소생술을 시행하였고, 심폐소생술 10분 경과 후에 자발적 순환 회복되었다.
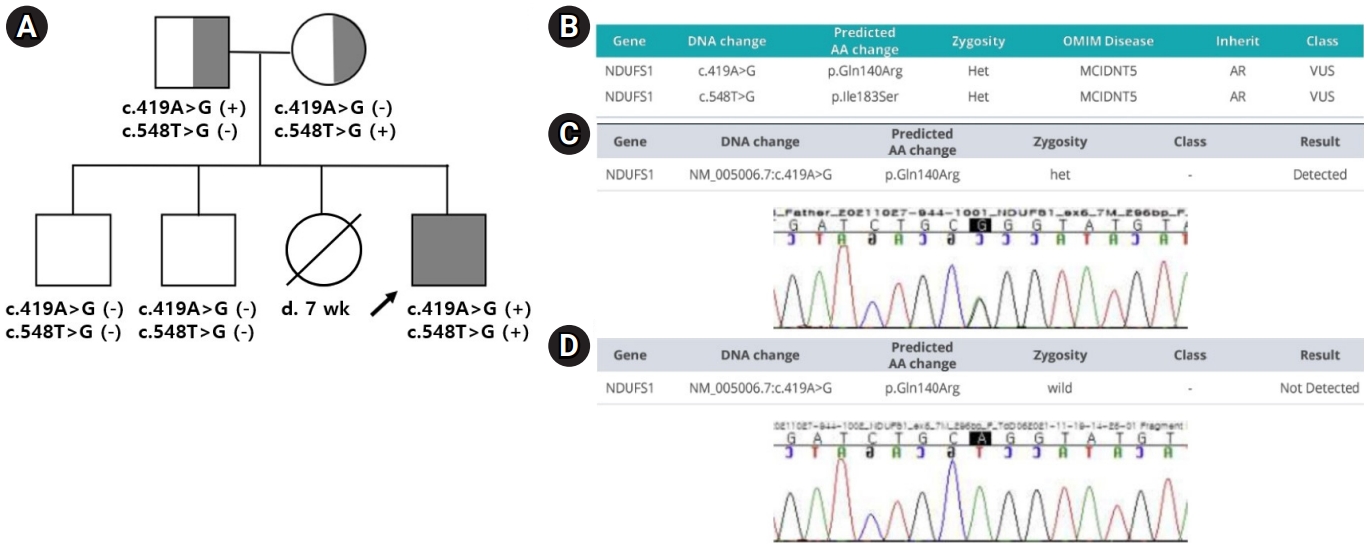
환아는 네번째 아이로, 재태 연령 36주 4일, 출생 체중 3,120 g으로 제왕절개로 태어났고, 내원 전까지 식이 및 성장은 정상이었다. 부모는 현재 신경학적인 이상 소견이나 유전적인 질환이 없었고, 10세와 6세의 두 아이는 정상적인 성장 및 발달을 보이고 있었다. 세번째 아이는 생후 46일 수면 중 영아 돌연사로 사망하였고, 당시 원인에 대한 다른 검사를 시행하지 않았다고 한다.
입원 1일째 혈액검사에서 혈청 lactate는 20.0 mmol/L(정상치 0.5~2.2 mmol/L), neuron specific enolase(NSE)는 44.2 ng/mL(정상치 0.0~16.3 ng/mL), 로 높았고, 입원 7일째 검사에서 lactate는 1.0 mmol/L으로 감소되었으나, NSE는 82.0 ng/mL으로 더 높게 증가되었다. 뇌파검사에서 양측 대뇌반구에서 극저전압 속파가 관찰된는 심한 뇌병증 소견을 보였다. 뇌자기공명영상 검사에서는 양측 기저핵과 시상, 중뇌의 깊은 회백질, 뇌량 및 해마 부위의 피질하 백질의 심한 뇌연화증 병변이 관찰되는 것으로, 리증후군의 특징적인 소견과 동반된 심한 저산소허혈성뇌병증(hypoxic-ischemic encephalopathy, HIE) 소견을 보였다(Fig. 1).
진단적 whole-exome sequencing(WES) 검사에서 미토콘드리아 호흡사슬의 complex I 기능을 저하시키는 것으로 알려져 있는, NDUFS1 유전자의 c.419A>G와 c.548T>G의 복합 이형접합 돌연변이를 확인할 수 있었다(참조서열: NM_005006.7). c419A>G와 c548T>G 변이는 한국인 참조 유전체 데이터베이스(Korean Reference Genome Database, KRGDB)와 genomAD에 발견된 적이 없는 매우 드문 변이이고(PM2), in-sillico prediction(SIFT, Polyphen2, Mutation Taster)에서 모두 유해하게 예측된 변이이다(PP3). 가족의 WES 검사 결과, 아버지는 NDUFS1 c.419A>G, p.Gln140Arg 이형접합 돌연변이를, 어머니는 NDUFS1 c.548T>G, p.Gln140Arg 이형접합 돌연변이를 가지고 있었고(PM3), 첫째와 둘째 아이는 돌연변이를 가지고 있지 않았다. 셋째 아이의 돌연사 소견(PP4)을 종합하여 American College of Medical Genetics and Genomics(ACMG) 가이드라인에 따라 두 변이를 ‘likely pathogenic’ 소견으로 판단하였다(Fig. 2).
환아는 인공호흡기를 이탈하지 못하여, 기관절개 및 가정용 인공호흡기를 사용하면서 퇴원하였고, 생후 8개월경 심초음파 검사에서 수축기 심실중격 두께 Z-score: 4.2-5.0, 수축기 좌심실 후벽 두께 Z-score: 5.1-6.1로 좌심실 근육 비대가 확인되었다. 이후 보존적 치료를 지속하던 중, 생후 10개월에 패혈증으로 사망하였다.
Discussion
리증후군은 (1) 신경퇴행성 증상, (2) 사립체 유전자의 돌연변이, 그리고 (3) 영상검사에서 양측성 중추신경계 이상 소견을 보이는 경우 진단하게 된다[1]. 리증후군의 임상증상은 그 발현 시기에 따라 신생아형, 영아형, 유년형으로 나눌 수 있는데, 자궁내에서부터 병리기전이 시작된다는 보고도 있다[6,7]. 전형적인 영아형의 신경퇴행성 증상은 2세 이전에 시작되는데, 가장 흔한 증상은 근육긴장감소, 강직, 운동실조, 무도증 등의 운동 이상증(83%)과 안구운동 이상(25%)이다[4]. 신생아형의 경우에는 수유장애, 무호흡, 입술청색증에서부터 심정지, 영아 돌연사와 같은 위중한 증상까지 아주 다양하게 나타난다[4,8]. 더욱이 신생아 시기에 증상을 보인 경우 뇌자기공명영상 검사나 혈액의 lactate가 정상을 보일 수 있어 조기 진단에 어려움을 준다[8].
본 증례에서도 심정지가 오기 전에 다른 신경학적인 증상을 보이지 않았고, 심정지로 인한 lactate 증가 및 HIE 소견으로 임상적 진단이 어려울 수 있었다. 하지만 셋째 아이의 영아 돌연사 가족력를 확인하고 바로 유전자검사를 시행하였고, WES 검사결과 NDUFS1 mutation을 확인할 수 있었다. 특히 환아는 아버지에서 발견된 NDUFS1 c419A>G, p.Gln140Arg 이형접합 돌연변이와 어머니에서 발견된 NDUFS1 c548T>G, p.Gln140Arg 이형접합 돌연변이를 모두 보이고 있고, 이는 아직 국내에 보고되지 않은 새로운 변이이다.
리증후군의 병인이 되는 사립체 유전자의 돌연변이는 사립체 유전자(mitochondrial DNA, mtDNA)에서 약 30%, 그리고 나머지는 핵 유전체(nuclear genome, nDNA)에서 확인된다[9]. 현재까지 75개 이상의 유전자가 사립체 및 핵 유전체(nuclear genome)에서 확인되었고, 이렇게 다양한 유전변이의 확인은 유전적인 다양성이 높음을 시사한다[9]. 1951년 리증후군의 첫 사례가 보고되고, 1991년 리증후군의 첫 유전변이가 확인된 이후, 현재까지 next generation sequencing와 WES과 같은 유전진단기술의 발전으로 다양한 유전변이와 비특이적인 임상양상이 보고되고 있어, 리증후군의 스펙트럼이 점점 넓어지고 있다[9,10]. 리증후군의 유전변이는 OXPHOS 기전에 필요한 여러 효소(pyruvate dehydrogenase, complex I~ V, coenzyme Q10 등)의 기능을 저하시키게 되는데, 이 중 complex I 결핍이 가장 흔하다[9,10]. 본 증례에서 확인된 NDUFS1 mutation도 complex I 기능을 저하시키는 원인 유전자로 잘 알려져 있다.
리증후군의 생존율은 15세에 68%, 16세에 57%, 20세에 20%정도로, 주로 2~3세에 사망하는 것으로 보고되고 있다[4,11,12]. 생후 6개월 이전에 증상이 발현되거나, 성장 지연이 있을 때, 뇌간에 영상학적 이상소견을 보일 때, 그리고 심기능장애를 보일 때 나쁜 예후를 보인다[4,12]. 현재까지 명확한 치료 방법은 없다. 리증후군을 포함한 OXPHOS 장애를 보이는 사립체질환에서 coenzyme Q10, L-carnitine, α-lipoic acid, creatine-monohydrate, biotin, thiamine, riboflavin 등의 약을 사용해 볼 수 있다[1]. 또한 multivitamin cocktail, ketogenic diet 등과 다양한 보존적인 치료방법을 시도하고 있지만, 결과는 만족스럽지 않다[3,4]. 본 증례도 자가용 인공호흡기 보조, 경관 식이, biotin과 thiamine 등의 보존적인 치료를 시행하였지만, 생후 10개월에 사망하였다. 이러한 나쁜 예후는 심정지로 시작된 이른 증상과, 심근비대의 심기능장애가 동반되었기 때문으로 생각된다.
본 증례는 이전에 신경학적인 증상없이 심정지를 첫 증상으로 내원하여 리증후군으로 진단된 경우로, 유전자 검사에서 새로운 NDUFS1 mutation을 확인할 수 있었다. 이처럼 리증후군은 영유아 급사의 원인이 될 수 있는 위중한 질환으로, 적극적인 유전자 검사를 통해 영아 돌연사 증후군의 원인 규명에 도움을 줄 수 있을 것이다. 또한 추가적인 분자 결함을 확인하는 것이 줄기세포 유래 사립체 기증과 같은 완치 방법의 연구에 도움을 줄 수 있을 것이다.